



高俊医生的科普号
- 精选 高颈段脊髓内肿瘤完全切除(续)
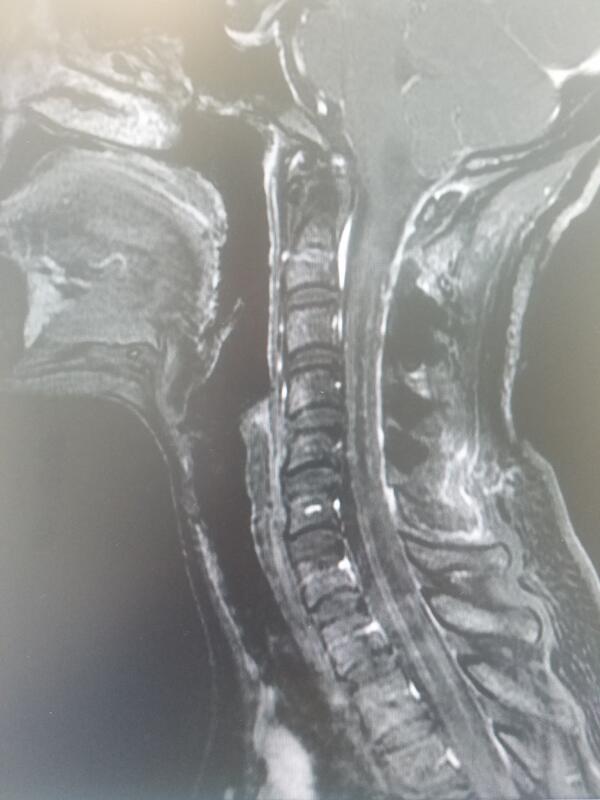
术后一周患者复查核磁显示肿瘤全切,复查CT椎板还纳位置满意,活动度和稳定性兼顾
 高俊 主任医师 北京协和医院 神经外科2488人已读
高俊 主任医师 北京协和医院 神经外科2488人已读 - 精选 高颈段脊髓内肿瘤完全切除(待续)
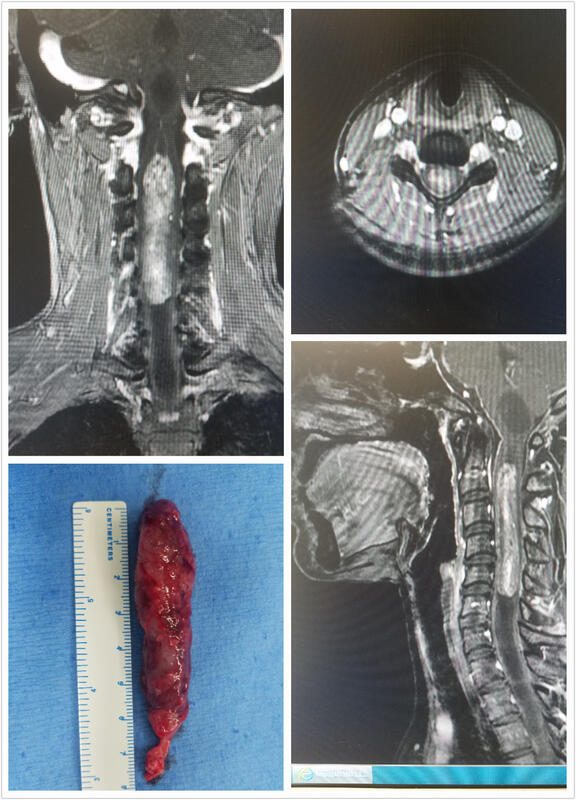
挑战神外手术禁区,切除7cm髓内肿瘤电生理监测下,历时3小时完全切除髓内肿瘤,术后患者四肢活动自如。一周内复查核磁后再上图片
 高俊 主任医师 北京协和医院 神经外科2849人已读
高俊 主任医师 北京协和医院 神经外科2849人已读 - 精选 半侧椎板入路微创切除椎管内肿瘤
胸椎管内先天性肿瘤位于脊髓的腹侧,将脊髓压迫至左侧椎管。术前患者双下肢的麻木无力进行性加重。手术采用半侧椎板入路,完全切除腹侧的肿瘤,解除脊髓的压迫。术中仅仅磨除右侧部分椎板,保留右侧小关节完整性;同时,对于左侧椎板、棘突、韧带及左侧椎旁肌肉无任何损伤,对于脊柱的稳定性基本上没有干扰,真正做到椎管内肿瘤的微创治疗。术后患者症状消失,第二天佩戴支具、下地行走。
高俊 主任医师 北京协和医院 神经外科1869人已读 - 精选 Arnold-Chiari畸形的研究进展
Chiari 畸形最早由奥地利医师Hans Chiari 在1891 年描述并命名,按照Chiari 畸形的解剖形态特将其分为4 型:I型为小脑扁桃体以及小脑蚓部疝入椎管内,但第四脑室保持在枕骨大孔以上;II型为第四脑室疝入椎管内;II型在I型或Ⅱ型的基础上,合并脊柱裂、脊膜膨出;IV型为小脑发育不全。其中以I型最为常见。过去,该疾病曾被认为是少见病,但是随着磁共振检查(MRI)的广泛应用,更多症状隐匿的患者被诊治和报道。20世纪末,Leslie等运用核磁共振检查,对美国北加利福尼亚20岁以下青少年做了一次随机抽样调查,在5248名青少年样本中,发现有55名患有Chiari畸形,发病率为1%。2003年,一项流行病学研究结果显示美国约有215,000名Chiari I型畸形患者。虽然国内尚无Chiari I型畸形患者的流行病学资料,但是由于庞大的人口基数,估计罹患该疾病的患者可达千万。近年来大量研究显示Chiari I型畸形具有很强的遗传倾向。Milhorat等对364名患者进行了家系调查,发现43名患者的近亲中至少还有一名以上的Chiari I型畸形患者。此外,文献报道中已有至少23个Chiari I型畸形家族。Chiari I型畸形患者的孪生兄弟/姐妹的发病几率明显增高,同卵双生同胞几乎100%发病。Chiari I型患者还多伴发其它明确的遗传性疾病,比如Klippel-Feil综合征等。因此,在基因组水平寻找Chiari I型畸形的疾病易感基因,并最终阐明该病的病因和发病机制,已成为Chiari I型畸形研究的热点。揭露Chiari I型畸形的基因背景,不仅对该疾病的诊断和治疗有着推动作用,对更好的了解其它类型的枕颈部发育畸形也是有积极意义的。近年来,科研人员对这方面做了有益的探索。Clare等认为Hox基因家族的Mhox基因调控枕骨的发育,在小鼠的Hox2.3基因变异可以导致枕骨发育不良,出现类似Chiari畸形的表型。Gripp等发现原癌基因HARS与Chiari畸形相关,HARS基因变异使细胞生长和增殖紊乱,导致脑和脊髓组织的过度生长,临床有报道此基因变异出现Chiari畸形的后颅凹发育缺陷、脊髓空洞、脑室扩大和小脑扁桃体下疝,并伴有巨头综合征。Wojcik等认为基因Suz12多梳蛋白表达异常可以导致脊柱裂、小脑及脑干畸形和脑积水,研究人员利用敲除Suz12杂合子成功建立了小鼠的Chiari畸形模型。尤其引人注意的是Boyles等对一批Chiari畸形患者及其家属的MRI作了研究,发现颅骨结构异常引起的后颅窝相对减小具有显著的遗传性(0.955, P=0.003),这一发现极大地支持了后颅凹容积减小的理论及其背后的基因遗传学基础。另外,对Boyles等还对23个家庭的71例Chiari畸形患者进行了全基因组连锁检测,测定了10000多个SNPs,以期界定是否与CMI的发生发展有关。结果显示,在15号染色体上有两个SNP的LOD值达到了3.3。多点测定发现存在一段13 CM的区域((15q21.1-22.3),其LOD值超过1。有趣的是,这段区域恰好包括一个生物学上可能与CMI高度有关的基因fibrillin-1(FBN1)。至今发现的FBN1基因突变已经有600多种,分布于整个FBN1基因,其中62种基因突变重复出现。根据迄今为止对FBN1基因及Fib-1蛋白的研究成果,已经基本明确其基因和蛋白的结构和功能。FBN1基因全长约235kb,由65个外显子构成,定位于15q21.1。其mRNA包含9663个核苷酸,由5'端非翻译区、开放读码框架和3'非翻译区组成,长度分别为134、8613和916个核苷酸。Fib-1蛋白由成纤维细胞和平滑肌细胞合成,由2871个氨基酸组成。该蛋白含有三种富含半胱氨酸的重复基序:第一种为类表皮生长因子基序;第二种为潜在的转化生长因子β1结合蛋白基序;第三种重复基序是Fib基序,是FBN1所特有的一种杂交基序,由EGF基序和TB基序融合而成。FBN1是构成细胞外微纤维(直径10~12nm)的主要蛋白之一,而微纤维可以作为弹性蛋白沉积及弹性纤维形成的股价,在组织中提供弹性支持作用,因此FBN1基因可以影响微纤维的结构和功能。该基因已被证实与马凡综合征和Shprintzen–Goldberg综合征高度相关,而Chiari畸形恰恰是许多伴随畸形的综合征尤其是Shprintzen–Goldberg综合征的特征性表现。但由于并非所有的Shprintzen–Goldberg综合征患者都伴随Chiari及畸形,也并非所有的Goldberg综合征患者都与FBN1有关,因此,位于15q21.1的FBN1基因的SNP究竟在Chiari畸形的发生发展中起多大的作用,仍然不甚明确。但无论如何,FBN1是一个值得探索的基因,也是值得在Chiari畸形患者及其家属细胞中深入考究并统计分析的基因。
高俊 主任医师 北京协和医院 神经外科3805人已读 - 精选 脊柱侧弯合并椎管内肿瘤
一名29岁男性患者,发现脊柱侧弯16年,偶尔感觉腰痛、双下肢乏力,未予以重视。此次入我院拟行手术矫形,术前检查行MRI发现椎管内占位。由于脊柱发育畸形,不仅存在侧凸,还伴有旋转,切除肿瘤时损伤脊髓的风险明显增大。我科脊柱脊髓专业组通过精确定位、显微操作,完整切除椎管内肿瘤(病理为支气管源性囊肿),术后未出现神经功能损伤症状;恢复3天后转入骨科行矫形手术,并如期出院。患者感觉不适症状明显缓解。
高俊 主任医师 北京协和医院 神经外科2178人已读 - 精选 腰椎管内神经鞘瘤切除
患者因“双下肢麻木不适一月”入院,行后路L3-L4椎管内肿瘤切除术,病理回报:神经鞘瘤。术后患者不适症状消失,术后1周出院
高俊 主任医师 北京协和医院 神经外科3476人已读 - 精选 高颈段哑铃型神经鞘瘤的显微外科治疗
主诉:右手麻木1年,加重伴双下肢无力5月余手术方式:基础远外侧入路C1、2神经鞘瘤切除术出院诊断:C1、2哑铃型神经鞘瘤康复情况:出院时不适症状明显改善,术后一个月患者正常工作生活
高俊 主任医师 北京协和医院 神经外科2143人已读 - 医学科普 内镜手术治疗Chiari I型畸形伴脊髓空洞症
关于小脑扁桃体下疝畸形的发病机制,比较公认的理论是:枕骨的发育不良导致先天性后颅窝容积偏小,而正常发育的后脑部分由于过度拥挤,使得小脑扁桃体下蚓部疝入到椎管内。尽管存在多种学说解释脊髓空洞积水的发病机制,其核心内容都是:小脑扁桃体下疝导致颅腔和椎管内的脑脊液循环受阻,进而引起脊髓中央管的扩大、积水。外科手术是治疗合并脊髓空洞积水的Chiari I型畸形的唯一有效措施。手术的关键步骤是彻底解除枕大孔区的狭窄、适当扩大后颅窝的容积。近些年,较小的骨窗(甚至1.5cm×2cm)也被证实能获得良好的减压效果,使得采用微创技术治疗脊髓空洞积水的Chiari I型畸形成为可能。 北京协和医院神经外科脊髓、脊柱专业组应用神经内镜照明、观察术野,内镜鞘外操作进行寰枕减压手术治疗伴有脊髓空洞症的I型畸形,手术效果满意。所有患者术后恢复顺利,无中枢神经系统感染、枕部皮下积液等并发症发生,无手术死亡病例。术后住院时间为3~14 天,平均住院时间6 天;住院费用在3.5万元左右。术后随访患者,全部患者均有改善。随诊时复查MRI,大部分患者的脊髓空洞较前不同程度缩小。图1 神经内镜下寰枕减压的手术切口。枕下发际处正中切口(以枕骨大孔为中心),约3cm。图2 一名32岁的男性患者主诉“枕颈部间断性疼痛4年,双上肢无力5个月”,完善检查后辅以神经内镜进行寰枕减压手术,术后第二天枕颈部不适明显缓解。术后复查MRI,显示枕大池容积较术前增大,颈髓内空洞较术前明显缩小。A、术前颈髓矢状位MRI;B、术后颈髓矢状位MRI。AB
高俊 主任医师 北京协和医院 神经外科7125人已读 - 典型病例 髓内畸胎瘤切除
男性,45岁,因“肢体麻木、无力进行性加重7年余 ”入院诊断:C2-5节段椎管内占位病变性质待查 髓内病变可能 神经电生理监测下C2-5髓内占位内病变探查切除术 术后病理:髓内占位病变符合成熟性囊性畸胎瘤
高俊 主任医师 北京协和医院 神经外科4606人已读 - 典型病例 髓内室管膜瘤切除
女性,26岁,因“颈枕部间断疼痛不适2月,双侧前臂尺侧麻木1月余 ”入院术前诊断:C6-T2髓内占位病变性质待查 胶质瘤可能行后正中入路髓内肿瘤切除术,术中全切肿瘤(长约7cm)术后病理:室管膜瘤 术后患者康复顺利,症状明显缓解
高俊 主任医师 北京协和医院 神经外科3900人已读

高俊主任医师
北京协和医院神经外科
